
In the late 19th century, a 32-year-old woman with an enlarged spleen came to attention of a French physician, named Philippe Gaucher. Unfortunately, the patient died of septicaemia. In his thesis, dr. Gaucher described the peculiar pathology of a hitherto unknown disorder, involving accumulation of large cells with striated cytoplasm that affected the spleen as well as the liver and seemed to be a non-malignant proliferation [1]. In the years thereafter, the clinical picture of Gaucher disease was described in more detail, including the familial pattern of appearance. Also, sub-types affecting the central nervous system, with variable involvement of the liver and spleen were identified. It took several years before the biochemical basis of Gaucher disease was unravelled. The identification of an accumulated glycosphingolipid, glucocerebroside, in the spleen cells eventually led to the discovery of the enzyme responsible to cleave the glucose from ceramide by Patrick and Brady in 1965. Deficiency of the enzyme glucocerebrosidase (also known as acid β-glucosidase or E.C.3.2.1.45) is the basis for the occurrence of glucocerebroside storage in macrophages. Many mutations have so far been identified at the gene for human glucocerebrosidase (GBA), which has been mapped to chromosome 1q21.
- CLINICAL FEATURES
- GENETICS/ INHERITANCE
- DIAGNOSIS
- PATHOPHYSIOLOGY
- THERAPY
- CLINICAL MONITORING INCLUDING RADIOLOGY
- EDUCATION
- GAUCHER CELL
COMING SOON
A diagnosis of Gaucher disease is usually based upon histological or cytological examination of bone marrow specimens, a liver biopsy, or a surgically removed spleen. The presence of lipid-laden macrophages with striated cytoplasmic inclusions, the Gaucher-cells, is not specific for the disease. Many other diseases, including chronic myeloproliferative diseases, malignancies, and chronic inflammatory disorders, have been described as giving rise to so-called ‘pseudo-Gaucher’ cells. For a reliable diagnosis of Gaucher disease, histological examinations are neither necessary nor sufficient. The detection of low glucocerebrosidase levels in leukocytes or urine is pathognomonic of Gaucher disease and should therefore be applied in cases of suspected Gaucher disease. However, since the disorder is rare and its clinical manifestations may mimic lymphoma or other haematological diseases, a bone marrow or liver biopsy will usually be performed before a diagnosis of Gaucher disease is even considered, especially in the absence of known affected family members. For the detection of carrier status, enzyme assays have limited value because of the considerable overlap with normal subjects. In these cases, mutation analysis is warranted.
COMING SOON
Palliative treatment
Before enzyme replacement therapy became available in the early 1990s, the treatment of type 1 Gaucher disease was merely palliative. Massive splenomegaly, causing severe anaemia and thrombocytopenia or leading to mechanical complaints, sometimes necessitated removal of the spleen. This almost invariably led to complete haematological recovery, especially of platelet counts. Whether splenectomy results in progressive storage of glycolipids at other sites, such as the liver, lung and bone marrow, has been subject to debate. Partial splenectomy has also been attempted to avoid the removal of the spleen as a major storage site, but splenic regrowth and surgical complications abandoned its use. In the majority of cases, splenectomy can be avoided with the institution of enzyme therapy. In rare instances, e.g. the presence of severe fibrosis, enzyme therapy may fail to reduce the size of the spleen, and splenectomy for persistent hypersplenism may still be necessary. For the palliative treatment of bone crises, analgesic drugs and bed rest are usually indicated. Bacterial osteomyelitis, which can be difficult to distinguish from bone crises, requires lengthy treatments with intravenous antibiotics. Orthopaedic procedures are sometimes indicated in cases of avascular necrosis or pathological fractures.
Enzyme replacement therapy
Early studies of enzyme replacement therapy involved either the intravenous infusion of small amounts of purified, unmodified glucocerebrosidase from placental tissue, or the administration of enzyme entrapped in liposomes or erythrocyte ghosts to facilitate macrophage uptake. The results of these studies were disappointing. However, improved targeting of the enzyme to macrophages was achieved (by modification of its glycosylation status and exposure of terminal mannose residues) and higher doses used. The first clinical trials with this new formulation showed a dramatic response in patients to regular intravenous infusion of this modified enzyme [1]. The industrial scale production of placenta-derived glucocerebrosidase and the development of the recombinant enzyme (CeredaseTM and CerezymeTM) made possible a number of further clinical trials, which confirmed the beneficial effects of enzyme therapy. Extremely high costs have made this treatment available only for those living in developed countries, and lower dosing regimens have thus been considered. Initial studies were performed with bi-weekly infusions of 60 U/kg per month. Other investigators advocated that lower dosages at higher frequencies were more efficient and therefore more cost-effective. In addition, home treatment, which further lowers costs, was shown to be feasible and more convenient for the patients [2]. In the Netherlands, a protocol was developed that allowed individualized treatment based upon each patient’s response, potentially providing an improved cost–benefit ratio [3]. In most patients, improvement in cytopenia and decreases in splenic and hepatic size are apparent after 3–12 months of treatment. Improvements in the haemoglobin level and especially in the platelet count are usually faster in splenectomised than in non-splenectomised patients. Liver volumes usually normalize, while some degree of splenic enlargement commonly persists, even after long-term treatment. Retarded growth in children and quality of life improves with treatment. Bone disease tends to respond much less rapidly than organomegaly to enzyme replacement therapy, but this depends largely on the sensitivity of the methods employed to assess the response. Many tools have been applied to assess organ responses to enzyme replacement. Plain X-rays are very insensitive and have no place in the assessment of treatment response; they should only be used in cases of bone complications. Magnetic resonance imaging is probably the best way to obtain information on bone marrow invasion and structure. Several scoring systems have been developed based upon changes in T1- and T2-weighted patterns. Quantitative chemical-shift imaging (QCSI) is the most sensitive method to assess bone marrow infiltration. This technique was further developed at the AMC by Mario Maas and Erik Akkerman and measures the ratio of triglyceride to water in the bone marrow [4]. In Gaucher disease patients, this ratio is greatly reduced, probably due to displacement of normal triglyceride-rich adipocytes in the bone marrow by Gaucher cells. AMC researchers have shown that the fat fraction of the bone marrow at the level of the lumbar spine relates to bone complications. Early reappearance of fatty marrow during enzyme replacement therapy can be detected with this technique. However, QCSI is not widely available and therefore limited in its use.
Despite the enormous success of enzyme therapy, several issues remain unresolved. For example, there is still no consensus about the criteria for initiation of treatment, the best way to monitor effects, and the most appropriate dosing regimen, particularly during the maintenance phase of treatment. The variability in clinical response to treatment and the extremely high costs (around $ 200 000 to 500 000 per patient per year) play an important role in these issues. As commitment to therapy is potentially life-long, the overall cost of care using enzyme therapy can be considerable, and very few reports are available about long-term therapeutic outcomes. Generally, therapeutic goals should be defined with the use of clinically relevant therapeutic endpoints, and protocols should be developed that aim to maintain an optimal effect while decreasing the burden of frequent infusions and the cost of care.
Substrate balance therapy
The accumulation of glucosylceramide is due to an imbalance between the rates of its synthesis and degradation. This concept forms the basis of a recently developed alternative oral therapy, termed substrate balance therapy (also known as substrate reduction therapy). Clearance of the accumulated glycolipid should be possible by attenuating the rate of synthesis of the substrate to a level that is matched to the residual activity of the endogenous, mutant glucosylceramidase. N-butyldeoxynojirimycin (OGT918 or miglustat) is an inhibitor of glucosylceramide synthesis, the first committed step in the biosynthesis of glycolipids. When tested in animal models of glycolipid storage disorders, the compound reduces the amount of storage material and delays the onset and progression of disease manifestations. Its mode of action suggests therapeutic potential in several of these diseases, including those with neurological involvement, since this small molecule is capable of crossing the blood–brain barrier. The first study in patients with a lysosomal storage disorder was initiated in 1998 in 28 type 1 Gaucher disease patients, who received 100 mg OGT918 orally three times daily [5]. The results of this study were promising: improvements occurred for all key clinical features and biochemical abnormalities. Liver and spleen volumes showed a gradual decrease in the first 6 months of treatment, while haematological improvement was slower. The most common adverse event was diarrhoea, which occurred in almost 90% of patients, especially during the first 3 months of treatment. Compared to enzyme therapy, the effects of OGT918 are slower to manifest. This and the disadvantageous side effects have to be balanced against the advantages of oral administration. Enzyme therapy remains the first choice for patients with moderate to severe disease, but mildly affected patients or those with minimal residual disease after treatment with enzyme may prefer this oral alternative. New oral alternatives are currently being developed.
COMING SOON
COMING SOON
In the late 19th century, a 32-year-old woman with an enlarged spleen came to the attention of a French physician, named Philippe Gaucher. Unfortunately, the patient died of septicemia. In his thesis, Dr. Gaucher described the unusual pathology of a hitherto unknown disorder, involving accumulation of large cells with striated cytoplasm that affected the spleen as well as the liver and seemed to be a non-malignant proliferation1. In the years thereafter, the clinical picture of Gaucher disease was described in more detail, including the familial pattern of appearance. Also, sub-types affecting the central nervous system, with variable involvement of the liver and spleen were identified. It took several years before the biochemical basis of Gaucher disease was unraveled. The identification of the accumulated glycosphingolipid, glucocerebroside, in splenic cells led Patrick and Brady to the discovery of the enzyme responsible for cleavage of glucose from ceramide in 1965 by. Deficiency of the enzyme glucocerebrosidase (also known as acid β-glucosidase, GCase) is the main basis for the occurrence of glucocerebroside accumulation in macrophages. Recently, enzyme misfolding and inflammation have also been suggested to contribute to the pathogenesis of Gaucher disease. Many mutations have so far been identified in the gene for human glucocerebrosidase (GBA1), which has been mapped to chromosome 1q21.
References:
- Gaucher P. De l’épithelioma primitif de la rate, hypertrophie idiopathique de la rate sans leukemie. [Thesis]. Paris, 1882.
Gaucher disease (GD) has traditionally been divided in three phenotypic variants. Type 1 GD is the most common variant and has a higher prevalence in the Ashkenazi Jewish population (frequency of 1:850)1. The overall frequency is believed to be around 1:50,000-1:100,000.
The clinical picture of type 1 GD is very heterogeneous: asymptomatic individuals have been identified during family studies and newborn screening, but very severe symptomatology at an early age can also occur. Splenomegaly is usually the first symptom, occurring in combination with thrombocytopenia and bleeding tendency2. In more advanced disease, the spleen can become grossly enlarged, filling the entire abdominal cavity. Splenic infarcts, which may be symptomatic or asymptomatic, occur. The splenic tissue is very firm and spontaneous rupture of the spleen rarely happens. Splenic consumption of peripheral blood cells results in cytopenias whilst reduction in coagulation factors by a number of means contributes to a bleeding tendency. With increasing spleen volume, platelets drop first, followed by erythrocytes and occasionally leucocytes. The combination of thrombocytopenia, platelet dysfunction and abnormally low clotting factors can result in severe bleeding. Infiltration of the bone marrow further contributes to the severity of cytopenia, in particular anaemia. Fatigue is a common complaint, which may be related to anemia, but also occurs in non-anemic patients and sometimes its chronicity and intensity are disabling. Gradual enlargement of the liver, with elevation of cholestatic liver enzymes, is usually found. After splenectomy, which was frequently performed in the past in severe cases of GD , the liver may become as grossly enlarged as the spleen, with end stage fibrosis as one of the most severe complications. Nowadays with the availability of therapy splenectomy is rarely required. High ferritin levels are commonly found in patients diagnosed with GD and may reflect macrophage activation and hepatic involvement.
Bone disease is a frequent manifestation of GD and is not always synchronous with visceral involvement3. Avascular necrosis of femoral heads may occur as the first manifestation of disease, even in the absence of palpable a spleen and liver. Bone complications include the occurrence of extremely painful bone crises, resembling sickle-cell crises, for which pain medications are indicated. Patients exhibit significant pain and elevation of inflammatory markers whilst blood cultures are sterile. Pathological fractures of the vertebrae and long bones can occur and not uncommonly osteopenia or osteoporosis can be found. The pathophysiology of bone disease is incompletely understood. With advanced storage in the bone marrow, secondary changes in bony architecture occur, leading to cortical thinning and marrow infarcts. Advanced bone disease can lead to requirement for joint replacement and severe impairments in mobility and quality of life.
Delayed menarche and menorrhagia are commonly described in untreated women with GD, and increased risk of recurrent abortions has been reported. Fertility is unaffected in males and females. When a female patient becomes pregnant, it is advisable that physicians experienced with GD be involved in follow-up with a multi-disciplinary approach to ensure safe pregnancy and delivery with particular attention to risk assessment of bleeding at the time of delivery.
Less frequent manifestations are parenchymal lung involvement and pulmonary hypertension. These manifestations usually occur only in advanced cases of GD 1 and in types 2 and 3 disease.
Patients with GD as well as GBA1 carriers are at increased risk of Parkinson disease. Considerable clinical variation is seen in GBA1-related Parkinson disease; some patients have early-onset and prominent cognitive changes while others have a later onset and slower course4.
Monoclonal gammopathy of undetermined significance (MGUS) has been associated with GD and the risk of multiple myeloma has been found to be more prevalent with an increased risk of 25- to 50-fold greater than expected in the general population5.
An association of GD with other hematological and solid cancer has also been described.
Type 2 and 3 GD are neuronopathic variants which are less commonly reported than type 1 but worldwide may have a significant incidence6. Type 2 disease is a very severe form, with rapidly fatal neurological symptomatology. Patients usually die at a young age. Type 3 disease is more variable, with predominance of visceromegaly or neurological symptomatology. Patients usually exhibit characteristic eye-movement abnormalities, with horizontal gaze palsy. Neurological symptoms can be minimal and remain stable for many years but can also progress to severe epilepsy and central neurological impairment. Type 3C has unique genetics (i.e., homozygosity to p.D448H (D409H) mutation) and unique phenotype including cardiac involvement.
Ideally, patients with GD should be managed by health care providers that have experience with the diverse clinical manifestations and potential GD related or unrelated co‐morbidities.
One of the EWGGD objectives is to enable education and support for physicians and health care providers with less experience in the field. Please contact us for more information.
References:
- Zimran A, Elstein D. Gaucher disease and related Lysosomal Storage Diseases. . In: K. K, Lichtman M, Beutler E et al., editors. Williams’ Hematology. New-York: McGraw-Hill; 2016.
- Mehta A, Kuter DJ, Salek SS , et al.: Presenting signs and patient co-variables in Gaucher disease: outcome of the Gaucher Early Diagnosis Consensus (GED-C) Delphi initiative. Intern Med J 2018; 49: 578-91.
- Hughes D, Mikosch P, Belmatoug N , et al.: Gaucher Disease in Bone: From Pathophysiology to Practice. J Bone Miner Res 2019; 34: 996-1031.
- Lopez G, Kim J, Wiggs E , et al.: Clinical course and prognosis in patients with Gaucher disease and parkinsonism. Neurol Genet 2016; 2:e57.
- Arends M, van Dussen L, Biegstraaten M, Hollak CE: Malignancies and monoclonal gammopathy in Gaucher disease; a systematic review of the literature. Br J Haematol. 161:832, 2013.
- Schiffmann R, Sevigny J, Rolfs A , et al.; The definition of neuronopathic Gaucher disease. J Inherit Metab Dis. 2020
- Gaucher cells, a major player in GD physiopathology
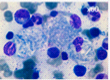
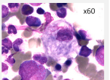
Mutations in the GBA1 gene lead to a marked decrease in β−glucocerebrosidase (GCase) activity that results in the accumulation of its substrate glucosylceramide, particularly in macrophages. The phagocytic role and naturally high level GCase activity of macrophages may partly explain why these cells are particularly affected in GD. Under light microscopy, Gaucher cells are typically enlarged, with eccentric nuclei and condensed chromatin. The cytoplasm has a heterogeneous “crumpled tissue paper” appearance (Figure 1), related to the presence of glucosylceramide aggregates in characteristic twisted, fibrillar arrangements that can be visualized by electron microscope1.
Gaucher cells mainly infiltrate bone marrow, spleen and liver, and are considered the main determinants of GD symptoms2.
Macrophages are cells of the innate immune system that adopt a variable phenotype and have different cellular properties depending on environmental stimuli. Although they display some plasticity, macrophages can be schematically distinguished in classical activated M1 macrophages and so-called alternative activated M2 macrophages. Not all monocytes/macrophages become Gaucher cells. Indeed, the subtype M2 (alternative differentiation pathway) appears to be particularly susceptible to GCase deficiency. This subset is characterized by anti-inflammatory, immunomodulatory and tissue repair properties, and includes macrophages that remove abnormal hematopoietic cells and phagocytose erythroblast nuclei. For this reason, some molecules synthesized by this macrophage subpopulation, such as chitotriosidase and CCL18, are considered to be biomarkers of Gaucher cell burden. Nevertheless, variable activation of M1 macrophages also has been observed, and may be responsible for the pseudo-inflammatory environment of GD, described several years ago.
Figure 1 : Gaucher cells in bone marrow (MGG staining)
2. Extended consequences of GCase deficiency, alteration of other cell types, relationship with co-morbidities
More recently, a number of studies have highlighted the consequences of GCase deficiency on cells other than macrophages. Although, major cytological changes are not observed in other cell types, functional changes have been described which demonstrate the much wider physiological of GCase deficiency.
2.1 Metabolic consequences besides glucosylceramide accumulation in Gaucher cells:
GCase deficiency leads to metabolic adaptations that probably have multiple and still poorly known consequences. For example, excess glucosylceramide is metabolized by lysosomal acid ceramidase into glucosylsphingosine (GlcSph) or lyso-GL1. GlcSph elevation in brain, spleen and plasma can be used as a biomarker of GD severity (see paragraph). In parallel, a compensatory increase in the activity of GCase GBA2, that works on the cytosolic side in neutral media, will promote the transformation of glucosylceramide into ceramide and of glucosylsphingosine into sphingosine-1-phosphate (S1P)3. Therefore, depending on the targeted tissue heterogeneity, these molecules may have some role in GD pathophysiology and determining severity.
2.2 Hematopoiesis:
It has been reported that GCase deficiency has a direct effect on the proliferation and differentiation capacities of hematopoietic progenitors and long-term culture initiating cells4,5. Although GCase deficiency effect on the megakaryocyte lineage is insignificant, in patients, thrombocytopenia is the most frequent cytopenia, suggesting a complex mechanism involving a number of factors, not least the role of splenomegaly. In parallel, the morphological, rheological and adhesive properties of mature red blood cells are altered in GD, related to the overexpression of basal cell adhesion molecule (Lu/BCAM)6, and improve under specific treatment7. These abnormalities demonstrate the multiple cellular consequences of GCase deficiency in hematopoietic tissue.
2.3 Immune system:
GCase deficiency has multiple effects on the immune system. For example, GD has been associated with a pro-inflammatory profile, impaired host-defense against microbial infections, upregulation of T-helper (Th)1 cells, and dysfunction of monocytes8-16.
Several mechanisms have been evoked to explain the immune system alterations. For example, CCL18 can recruit CCR8-expressing T regulatory cells and modify the immune microenvironment. In addition, CCL18 ability to recruit Th2 cells could help to maintain the M2 phenotype of macrophages and promote allergic reactions17-19.
Sphingosine-1-phosphate (S1P) is an important signaling molecule that can stimulate proliferation, motility, migration, and survival of many cell types, including lymphocytes and monocytes20. S1P induces pro-inflammatory M1 and anti-inflammatory M2 monocyte migration21,22, and could play a role in favouring the M2 or M1 phenotype of macrophages, depending on the tissue and eventual damage23,24. S1P also has a role in mediating naive T-cell egression from lymphoid organs25.
However, the clinical consequences of such immune dysfunction remain moderate, suggesting the existence of compensatory mechanisms and/or the need of prolonged disruption for clinical symptoms to occur. For instance, B cell stimulation results in an increased risk of polyclonal hypergammaglobulinemia and monoclonal gammopathy26,27, and a favorable environment for the emergence of B-cell malignancies, such as myeloma. Chronic sphingolipid excess may become an antigenic target that stimulates B cells and promotes the development of monoclonal gammopathies and then myeloma, although the exact antigenic target remains debated28-31.
Overall, these immune system changes could contribute to the mechanisms that promote the emergence of cancers in GD (see Cancer)
2.4 Bone tissue:
Bone changes are observed in the vast majority of patients, but with very heterogeneous clinical expression (see clinical symptomatology). It is commonly accepted that bone marrow infiltration visualized by MRI and a deformity of the distal femur in the shape of an Erlenmeyer flask are related to the infiltration of the bone marrow cavity by Gaucher cells, especially in the period of tissue growth. However, the exact role of Gaucher cells or other macrophages and related cells remains unknown. Most bone pathology suggests an altered balance between synthesis (osteoblasts) and resorption (osteoclasts) of bone tissue. However, studies of bone metabolism biomarkers have given contradictory results32.
Several studies have demonstrated that GCase deficiency promotes the abnormal proliferation of mesenchymal stem cells (MSCs), a major actor in bone homeostasis. GD-MSCs display impaired growth potential, morphological and cell cycle abnormalities, and decreased capacity to differentiate into osteoblasts. Moreover, GD-MSCs secrete soluble factors that stimulate the osteoclast resorbing activities33. Similarly, the properties of mesenchymal cells are altered. Metabolites produced by the GBA2 pathway, such as sphingosine, could have a direct toxic effect on bone tissue and particularly on osteoblasts34. Other studies have shown a predisposition of monocytic cells to differentiate into osteoclasts and a reduced ability to mineralize the bone matrix35. However, the contribution of these different perturbations on bone damage in GD, particularly the occurrence of bone infarction (osteonecrosis) remains to be clarified.
2.5 Neurological impairment
Type 2 and type 3 GD are characterized by neurological damage, but long-term monitoring of type 1 GD has shown the presence of neurological injuries with higher risk of Parkinson’s disease and Lewy body dementia also in this variant.
Neurological impairment in type 2 and 3 GD:
The pathophysiological mechanisms of neurological impairment in GD remain poorly explained. Cells that resemble Gaucher cells have been observed in the brain of patients with type 2 and type 3 GD36–42. In brain, Gaucher cell infiltration is clearly less important than in bone marrow or spleen, and the origin of these cells remains unknown. Loss of neuronal cells and astrocytes also has been described37,41. Microglial cells, which can be considered the equivalent of macrophages in the brain, are probably involved, but more evidence is needed. Microglial cells are sensitive to the local inflammatory context, as reported several times, and might be actively involved in brain inflammation, notably through the secretion of sphingosine-1-phosphate (S1P) linked to the accumulation of glucocerebroside and glucosylsphingosine in the brain. More recently, alterations in autophagy have also been demonstrated43. Better understanding the pathophysiology of nerve damage remains a major challenge in GD.
Relationship between the GBA1 gene and Parkinson’s Disease (PD)
The strong relationship between mutations in GBA1, the gene encoding GCase, and PD has been demonstrated by the observation that patients with GD are at higher risk of developing PD, and also by the higher prevalence of heterozygous GBA1 mutations (carriers) among patients with PD44. Moreover, GBA1 mutations are the genetic abnormality most frequently associated with PD. Several groups have attempted to determine the underlying pathophysiological mechanisms of this relationship.
Aggregated α-synuclein and Lewy bodies are observed in brain tissue of patients with GD45. Data supporting a role of the loss of GCase function as well as a gain of toxic function of mutated GCase in promoting α-synuclein accumulation have been published46.
Loss of GCase function leads to the accumulation of glucosylceramide and glucosylsphingosine. This hampers lysosomal α-synuclein degradation by decreasing the activity of the lysosomal aspartyl protease cathepsin D, thus stabilizing the insoluble toxic α-synuclein oligomeric intermediates and promoting the formation of α-synuclein aggregates47. This results in neurotoxicity through α-synuclein accumulation in the substantia nigra of the brain. Glucosylceramide, the GCase substrate, directly influences the formation of α-synuclein amyloid fibrils by stabilizing soluble oligomers that then aggregate and form Lewy bodies in nerve cells in PD. In addition, a vicious circle may occur since α-synuclein polymers can inhibit GCase activity48–50. Autophagy also has been involved in the pathophysiology of both GD and PD. Indeed, GCase deficiency impairs autophagy, thus promoting α-synuclein accumulation50,51.
The contribution of a gain of toxic function of mutated GCase on α-synuclein accumulation and aggregation has been recently demonstrated. Indeed, heterologous expression of mutated GCase in human neuroblastoma cells leads to a significant stabilization of α-synuclein and to its aggregation. Furthermore, in models of Drosophila melanogaster co-expressing human α-synuclein and human mutant GCase in the dopaminergic cells initiated α-synuclein aggregation and resulted in earlier death and significantly shorter life span, compared with flies expressing α-synuclein or mutant GCase alone52.
2.6 Relationship between GCase deficiency and cancer
The risk of developing specific types of cancer is higher in patients with GD than in the general population. Changes in the immune system appear to be a major determinant of the increased risk of myeloma in patients with GD (see above). However, other alterations that promote immunotolerance, such as increased activation of M2 macrophages that are similar to tumour-associated macrophages (TAM)53, increase in Th2 lymphocytes, or reduction of natural killer (NK cells), also may contribute to the higher cancer risk. Moreover, the chronic pseudo-inflammatory environment linked to GD might promote carcinogenesis. Furthermore, in cancer, the balance between ceramide and S1P signaling is important for regulating cell survival, cycling, and invasion. In GD, a shift toward S1P accumulation leads to pro-survival, anti-apoptotic, metastatic and drug-resistance phenotypes54,55.
References:
- Lee RE. The fine structure of the cerebroside occurring in Gaucher’s disease. Proc. Natl. Acad. Sci. U.S.A. 1968;61(2):484–489.
- Mikosch P, Hughes D. An overview on bone manifestations in Gaucher disease. Wien Med Wochenschr. 2010;160(23–24):609–624.
- Chipeaux C, de Person M, Burguet N, et al. Optimization of ultra-high pressure liquid chromatography – tandem mass spectrometry determination in plasma and red blood cells of four sphingolipids and their evaluation as biomarker candidates of Gaucher’s disease. J Chromatogr A. 2017;1525:116–125.
- Berger J, Lecourt S, Vanneaux V, et al. Glucocerebrosidase deficiency dramatically impairs human bone marrow haematopoiesis in an in vitro model of Gaucher disease. Br. J. Haematol. 2010;150(1):93–101.
- Reihani N, Arlet J-B, Dussiot M, et al. Unexpected macrophage-independent dyserythropoiesis in Gaucher disease. Haematologica. 2016;101(12):1489–1498.
- Franco M, Collec E, Connes P, et al. Abnormal properties of red blood cells suggest a role in the pathophysiology of Gaucher disease. Blood. 2013;121(3):546–555.
- Franco M, Reihani N, Marin M, et al. Effect of velaglucerase alfa enzyme replacement therapy on red blood cell properties in Gaucher disease. Am. J. Hematol. 2017;92(9):E561–E563.
- Allen MJ, Myer BJ, Khokher AM, Rushton N, Cox TM. Pro-inflammatory cytokines and the pathogenesis of Gaucher’s disease: increased release of interleukin-6 and interleukin-10. QJM. 1997;90(1):19–25.
- Lacerda L, Arosa FA, Lacerda R, et al. T cell numbers relate to bone involvement in Gaucher disease. Blood Cells Mol. Dis. 1999;25(2):130–138.
- Balreira A, Lacerda L, Miranda CS, Arosa FA. Evidence for a link between sphingolipid metabolism and expression of CD1d and MHC-class II: monocytes from Gaucher disease patients as a model. Br. J. Haematol. 2005;129(5):667–676.
- Mizukami H, Mi Y, Wada R, et al. Systemic inflammation in glucocerebrosidase-deficient mice with minimal glucosylceramide storage. J. Clin. Invest. 2002;109(9):1215–1221.
- Maródi L, Káposzta R, Tóth J, László A. Impaired microbicidal capacity of mononuclear phagocytes from patients with type I Gaucher disease: partial correction by enzyme replacement therapy. Blood. 1995;86(12):4645–4649.
- Finkelstein R, Nachum Z, Reissman P, et al. Anaerobic osteomyelitis in patients with Gaucher’s disease. Clin. Infect. Dis. 1992;15(5):771–773.
- Barak V, Acker M, Nisman B, et al. Cytokines in Gaucher’s disease. Eur. Cytokine Netw. 1999;10(2):205–210.
- Lichtenstein M, Zimran A, Horowitz M. Cytokine mRNA in Gaucher disease. Blood Cells Mol. Dis. 1997;23(3):395–401.
- Liel Y, Rudich A, Nagauker-Shriker O, Yermiyahu T, Levy R. Monocyte dysfunction in patients with Gaucher disease: evidence for interference of glucocerebroside with superoxide generation. Blood. 1994;83(9):2646–2653.
- Wei L, Vahedi G, Sun H-W, et al. Discrete roles of STAT4 and STAT6 transcription factors in tuning epigenetic modifications and transcription during T helper cell differentiation. Immunity. 2010;32(6):840–851.
- Islam SA, Chang DS, Colvin RA, et al. Mouse CCL8, a CCR8 agonist, promotes atopic dermatitis by recruiting IL-5+ T(H)2 cells. Nat. Immunol. 2011;12(2):167–177.
- Islam SA, Ling MF, Leung J, Shreffler WG, Luster AD. Identification of human CCR8 as a CCL18 receptor. J. Exp. Med. 2013;210(10):1889–1898.
- Maceyka M, Harikumar KB, Milstien S, Spiegel S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012;22(1):50–60.
- Awojoodu AO, Ogle ME, Sefcik LS, et al. Sphingosine 1-phosphate receptor 3 regulates recruitment of anti-inflammatory monocytes to microvessels during implant arteriogenesis. Proc. Natl. Acad. Sci. U.S.A. 2013;110(34):13785–13790.
- Weichand B, Weis N, Weigert A, et al. Apoptotic cells enhance sphingosine-1-phosphate receptor 1 dependent macrophage migration. Eur. J. Immunol. 2013;43(12):3306–3313.
- Park S-J, Lee K-P, Kang S, et al. Sphingosine 1-phosphate induced anti-atherogenic and atheroprotective M2 macrophage polarization through IL-4. Cell. Signal. 2014;26(10):2249–2258.
- Gaire BP, Bae YJ, Choi JW. S1P1 Regulates M1/M2 Polarization toward Brain Injury after Transient Focal Cerebral Ischemia. Biomol Ther (Seoul). 2019;522–529.
- Matloubian M, Lo CG, Cinamon G, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427(6972):355–360.
- Marti GE, Ryan ET, Papadopoulos NM, et al. Polyclonal B-cell lymphocytosis and hypergammaglobulinemia in patients with Gaucher disease. Am. J. Hematol. 1988;29(4):189–194.
- de Fost M, Out TA, de Wilde FA, et al. Immunoglobulin and free light chain abnormalities in Gaucher disease type I: data from an adult cohort of 63 patients and review of the literature. Ann. Hematol. 2008;87(6):439–449.
- Nair S, Boddupalli CS, Verma R, et al. Type II NKT-TFH cells against Gaucher lipids regulate B-cell immunity and inflammation. Blood. 2015;125(8):1256–1271.
- Nair S, Branagan AR, Liu J, et al. Clonal Immunoglobulin against Lysolipids in the Origin of Myeloma. N. Engl. J. Med. 2016;374(6):555–561.
- Preuss K-D, Hollak CEM, Fadle N, et al. Saposin C is a frequent target of paraproteins in Gaucher disease-associated MGUS/multiple myeloma. Br. J. Haematol. 2019;184(3):384–391.
- Nair S, Bar N, Xu ML, Dhodapkar M, Mistry PK. Glucosylsphingosine but not Saposin C, is the target antigen in Gaucher disease-associated gammopathy. Mol. Genet. Metab. 2020;129(4):286–291.
- van Dussen L, Lips P, Everts VE, et al. Markers of bone turnover in Gaucher disease: modeling the evolution of bone disease. J. Clin. Endocrinol. Metab. 2011;96(7):2194–2205.
- Campeau PM, Rafei M, Boivin M-N, et al. Characterization of Gaucher disease bone marrow mesenchymal stromal cells reveals an altered inflammatory secretome. Blood. 2009;114(15):3181–3190.
- Mistry PK, Liu J, Sun L, et al. Glucocerebrosidase 2 gene deletion rescues type 1 Gaucher disease. Proc. Natl. Acad. Sci. U.S.A. 2014;111(13):4934–4939.
- Reed MC, Schiffer C, Heales S, Mehta AB, Hughes DA. Impact of sphingolipids on osteoblast and osteoclast activity in Gaucher disease. Mol. Genet. Metab. 2018;124(4):278–286.
- Adachi M, Wallace BJ, Schneck L, Volk BW. Fine structure of central nervous system in early infantile Gaucher’s disease. Arch Pathol. 1967;83(6):513–526.
- Conradi NG, Sourander P, Nilsson O, Svennerholm L, Erikson A. Neuropathology of the Norrbottnian type of Gaucher disease. Morphological and biochemical studies. Acta Neuropathol. 1984;65(2):99–109.
- Conradi NG, Kalimo H, Sourander P. Reactions of vessel walls and brain parenchyma to the accumulation of Gaucher cells in the Norrbottnian type (type III) of Gaucher disease. Acta Neuropathol. 1988;75(4):385–390.
- Conradi N, Kyllerman M, Månsson JE, Percy AK, Svennerholm L. Late-infantile Gaucher disease in a child with myoclonus and bulbar signs: neuropathological and neurochemical findings. Acta Neuropathol. 1991;82(2):152–157.
- Kaye EM, Ullman MD, Wilson ER, Barranger JA. Type 2 and type 3 Gaucher disease: a morphological and biochemical study. Ann. Neurol. 1986;20(2):223–230.
- Wong K, Sidransky E, Verma A, et al. Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol. Genet. Metab. 2004;82(3):192–207.
- Yu Z, Sawkar AR, Whalen LJ, Wong C-H, Kelly JW. Isofagomine- and 2,5-anhydro-2,5-imino-D-glucitol-based glucocerebrosidase pharmacological chaperones for Gaucher disease intervention. J. Med. Chem. 2007;50(1):94–100.
- Kinghorn KJ, Asghari AM, Castillo-Quan JI. The emerging role of autophagic-lysosomal dysfunction in Gaucher disease and Parkinson’s disease. Neural Regen Res. 2017;12(3):380–384.
- Sidransky E, Nalls MA, Aasly JO, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 2009;361(17):1651–1661.
- Tayebi N, Walker J, Stubblefield B, et al. Gaucher disease with parkinsonian manifestations: does glucocerebrosidase deficiency contribute to a vulnerability to parkinsonism? Mol. Genet. Metab. 2003;79(2):104–109.
- Gegg ME, Schapira AHV. The role of glucocerebrosidase in Parkinson disease pathogenesis. FEBS J. 2018;285(19):3591-3603.
- Yang S-Y, Gegg M, Chau D, Schapira A. Glucocerebrosidase activity, cathepsin D and monomeric α-synuclein interactions in a stem cell derived neuronal model of a PD associated GBA1 mutation. Neurobiol. Dis. 2020;134:104620.
- Liu G, Boot B, Locascio JJ, et al. Specifically neuropathic Gaucher’s mutations accelerate cognitive decline in Parkinson’s. Ann. Neurol. 2016;80(5):674–685.
- Cilia R, Tunesi S, Marotta G, et al. Survival and dementia in GBA-associated Parkinson’s disease: The mutation matters. Ann. Neurol. 2016;80(5):662–673.
- Mazzulli JR, Xu Y-H, Sun Y, et al. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011;146(1):37–52.
- Pitcairn C, Wani WY, Mazzulli JR. Dysregulation of the autophagic-lysosomal pathway in Gaucher and Parkinson’s disease. Neurobiol. Dis. 2019;122:72–82.
- Maor G, Rapaport D, Horowitz M. The effect of mutant GBA1 on accumulation and aggregation of α-synuclein. Hum. Mol. Genet. 2019;28(11):1768–1781.
- Wątek M, Piktel E, Wollny T, et al. Defective Sphingolipids Metabolism and Tumor Associated Macrophages as the Possible Links Between Gaucher Disease and Blood Cancer Development. Int J Mol Sci. 2019;20(4):.
- Morad SAF, Cabot MC. Ceramide-orchestrated signalling in cancer cells. Nat. Rev. Cancer. 2013;13(1):51–65.
- Selvam SP, Ogretmen B. Sphingosine kinase/sphingosine 1-phosphate signaling in cancer therapeutics and drug resistance. Handb Exp Pharmacol. 2013;(216):3–27.
Although historically, the diagnosis of Gaucher disease (GD) has been historically made by detecting Gaucher cells on a bone marrow preparation, the identification of Gaucher cells can occasionally be problematic because it is influenced by their frequency and the quality of the bone marrow sample. Gaucher cells may also be confused with Gaucher-like cells (called pseudo-Gaucher cells) observed in other pathological conditions where cellular turnover is high for example in myeloproliferative conditions. In such cases an alternative mechanism of diagnosis is required.
For this reason, the detection of deficient GCase activity, usually in circulating leukocytes, is considered the essential diagnostic test. It must be performed by a specialist laboratory. Indications for testing include unexplained splenomegaly, pre-empting splenectomy performed for diagnostic purposes, and thrombocytopenia associated with hyperferritinemia. Case analysis can be performed using fresh blood or dried blood spot samples1, and a functional test based on a fluorescent substrate (4-methylumbelliferyl-β-D-glucopyranoside (4-MU-β-Glu)2–4. More recently a tandem mass spectrometry technique has been developed5. In patients with GD, GCase residual activity represents globally 10-15% of the activity measured in control subjects6–8.The possibility of carrying out this assay using dried blood spots (DBS) allows large-scale screening with very little and without the constraints of transporting fresh whole blood. However, the diagnosis should be confirmed by assessing the GCase activity in cells and/or by molecular analysis of GBA1 gene.
In very rare cases, if the clinical presentation is evocative but GCase activity is normal, molecular analysis of PSAP gene might be performed to identify GD caused by deficiency of saposin C, a molecule that promotes GCase activity.
Biomarkers:
The metabolic changes related to GCase deficiency induce the increased macrophage secretion or derivation from substrates of several molecules, some of which may be used as biomarkers of GD
Quantification of tartrate-resistant acid phosphatase and angiotensin converting enzyme levels has been used for many years, but these analysis lacks specificity and has become obsolete9. Currently, the main biomarkers used in clinical practice are chitotriosidase, CCL18/PARC, and glucosylsphingosine (lyso-Gb1).
Chitotriosidase and CCL18/PARC are two molecules synthesized by Gaucher cells10 and are considered as biomarkers of Gaucher cell burden, and thus indirectly of the disease severity. Glucosylsphingosine (or lysoglucosylceramide, lyso-GL1, lyso-Gb1) is the deacetylated derivative of glucosylceramide, and it is clearly increased in GD:
1) Chitotriosidase: Plasma chitotriosidase activity is strongly increased in GD, by about 1000 times the value of control subjects11,12. Plasma chitotriosidase activity quantification allows monitoring the treatment efficacy and seems to have some prognostic value13. However, chitotriosidase activity is highly variable in patients and also in the general population due to the presence of a possible mutation (duplication of 24 bp) in the CHIT1 gene that leads to a total deficit (mutation present in the homozygous state in 6% of individuals in the general Caucasian population) or partial deficit (30-40% with the heterozygous mutation). Therefore, it does not allow comparing patients and treatments14–16. Other polymorphisms might also influence the activity of chitotriosidase 17.
It must be noted that chitotriosidase activity can be increased also in other diseases involving macrophages, such as Niemann Pick disease, sarcoidosis, leishmaniasis and atherosclerosis.
2) CCL18/PARC (chemokine (C-C motif) ligand 18/pulmonary and activation-regulated chemokine): CCL18 is a chemokine produced by different cell types, particularly M2 macrophages. Gaucher cells also produce CCL18 and their plasma level is increased by a factor of 20-50 in patients compared with the general population18. Inter-individual variability is less important than that of chitotriosidase because of the absence of known genetic polymorphisms to date.
CCL18 may also be increased in Niemann Pick disease, sarcoidosis, pulmonary fibrosis, atherosclerosis, atopy/asthma, and cancers.
Chitotriosidase and CCL18 show similar reduction kinetics when the specific treatment is initiated and very similar performance in assessing the haemato-visceral severity of GD119. Their relationship with GD bone and neurological phenotypes is still debated.
3) Glucosylsphingosine (or lysoglucosylceramide, lyso-GL1, Lyso-Gb1): Although it was previously described as elevated in the neurological forms (type 2 and 3), the interest of lyso-GL1 as a biomarker has recently increased on the basis of several studies showing that lyso-GL1 i) is involved in immune dysregulation and skeletal diseases; ii) is widely diffused due to its solubility; iii) is increased in neurological tissues, and iv) is specifically increased in GD samples20,21. Therefore, lyso-GL1 is going to be considered a more specific biomarker with potentially better performance in assessing disease severity and prognosis than chitotriosidase and CCL1820,22,23. However, lyso-GL1, chitotriosidase and CCL18 changes during specific treatment are similar. The value of the different biomarkers may vary by phenotypic subgroups; further studies are needed to determine their respective performances.
References:
- Stroppiano M, Calevo MG, Corsolini F, et al. Validity of β-D-glucosidase activity measured in dried blood samples for detection of potential Gaucher disease patients. Clin. Biochem. 2014;47(13–14):1293–1296.
- Raghavan SS, Topol J, Kolodny EH. Leukocyte beta-glucosidase in homozygotes and heterozygotes for Gaucher disease. Am. J. Hum. Genet. 1980;32(2):158–173.
- Olivova P, Cullen E, Titlow M, et al. An improved high-throughput dried blood spot screening method for Gaucher disease. Clin. Chim. Acta. 2008;398(1–2):163–164.
- Chamoles NA, Blanco M, Gaggioli D, Casentini C. Gaucher and Niemann-Pick diseases–enzymatic diagnosis in dried blood spots on filter paper: retrospective diagnoses in newborn-screening cards. Clin. Chim. Acta. 2002;317(1–2):191–197.
- Wolf P, Alcalay RN, Liong C, et al. Tandem mass spectrometry assay of β-glucocerebrosidase activity in dried blood spots eliminates false positives detected in fluorescence assay. Mol. Genet. Metab. 2018;123(2):135–139.
- Neufeld EF. Lysosomal storage diseases. Annu. Rev. Biochem. 1991;60:257–280.
- Santos DM, Tiscornia G. Induced Pluripotent Stem Cell Modeling of Gaucher’s Disease: What Have We Learned? Int J Mol Sci. 2017;18(4):.
- Berger J, Vigan M, Pereira B, et al. Intra-monocyte Pharmacokinetics of Imiglucerase Supports a Possible Personalized Management of Gaucher Disease Type 1. Clin Pharmacokinet. 2019;58(4):469–482.
- Aerts JMFG, Kallemeijn WW, Wegdam W, et al. Biomarkers in the diagnosis of lysosomal storage disorders: proteins, lipids, and inhibodies. J. Inherit. Metab. Dis. 2011;34(3):605–619.
- Boven LA, van Meurs M, Boot RG, et al. Gaucher cells demonstrate a distinct macrophage phenotype and resemble alternatively activated macrophages. Am. J. Clin. Pathol. 2004;122(3):359–369.
- Hollak CE, van Weely S, van Oers MH, Aerts JM. Marked elevation of plasma chitotriosidase activity. A novel hallmark of Gaucher disease. J. Clin. Invest. 1994;93(3):1288–1292.
- Bussink AP, van Eijk M, Renkema GH, Aerts JM, Boot RG. The biology of the Gaucher cell: the cradle of human chitinases. Int. Rev. Cytol. 2006;252:71–128.
- van Dussen L, Hendriks EJ, Groener JEM, et al. Value of plasma chitotriosidase to assess non-neuronopathic Gaucher disease severity and progression in the era of enzyme replacement therapy. J. Inherit. Metab. Dis. 2014;37(6):991–1001.
- Boot RG, Renkema GH, Verhoek M, et al. The human chitotriosidase gene. Nature of inherited enzyme deficiency. J. Biol. Chem. 1998;273(40):25680–25685.
- Grace ME, Balwani M, Nazarenko I, Prakash-Cheng A, Desnick RJ. Type 1 Gaucher disease: null and hypomorphic novel chitotriosidase mutations-implications for diagnosis and therapeutic monitoring. Hum. Mutat. 2007;28(9):866–873.
- Bussink AP, Verhoek M, Vreede J, et al. Common G102S polymorphism in chitotriosidase differentially affects activity towards 4-methylumbelliferyl substrates. FEBS J. 2009;276(19):5678–5688.
- Irún P, Alfonso P, Aznarez S, Giraldo P, Pocovi M. Chitotriosidase variants in patients with Gaucher disease. Implications for diagnosis and therapeutic monitoring. Clin. Biochem. 2013;46(18):1804–1807.
- Boot RG, Verhoek M, de Fost M, et al. Marked elevation of the chemokine CCL18/PARC in Gaucher disease: a novel surrogate marker for assessing therapeutic intervention. Blood. 2004;103(1):33–39.
- Raskovalova T, Deegan PB, Mistry PK, et al. Accuracy of chitotriosidase activity and CCL18 concentration in assessing type I Gaucher disease severity. A systematic review with meta-analysis of individual participant data. Haematologica. 2020;
- Rolfs A, Giese A-K, Grittner U, et al. Glucosylsphingosine is a highly sensitive and specific biomarker for primary diagnostic and follow-up monitoring in Gaucher disease in a non-Jewish, Caucasian cohort of Gaucher disease patients. PLoS ONE. 2013;8(11):e79732.
- Pettazzoni M, Froissart R, Pagan C, et al. LC-MS/MS multiplex analysis of lysosphingolipids in plasma and amniotic fluid: A novel tool for the screening of sphingolipidoses and Niemann-Pick type C disease. PLoS ONE. 2017;12(7):e0181700.
- Dekker N, van Dussen L, Hollak CEM, et al. Elevated plasma glucosylsphingosine in Gaucher disease: relation to phenotype, storage cell markers, and therapeutic response. Blood. 2011;118(16):e118-127.
- Mirzaian M, Wisse P, Ferraz MJ, et al. Mass spectrometric quantification of glucosylsphingosine in plasma and urine of type 1 Gaucher patients using an isotope standard. Blood Cells Mol. Dis. 2015;54(4):307–314.
Molecular diagnosis
Gaucher disease (GD) is caused by biallelic pathogenetic mutations in the gene encoding the GCase enzyme, GBA1 (GRCh37/hg19 Chromosome 1: 155,204,239 to 155,214,653). However, very rare cases are due to biallelic mutation in the PSAP gene (NM_002778.4), leading to the specific deficiency of the GCase activator protein, Saposin C.
The GBA1 gene encompasses 11 coding exons spanning approximately 7.6 kb of sequence, while the cDNA is approximately 2.5 kb. Two in-frame ATG translational sites located in exons 1 and 2 have been described; both are efficiently used to generate two polypeptides with signal peptides of 39 and 19 residues, respectively.
The gene has been located to chromosome 1q21 within a complex locus containing seven genes and two pseudogenes, likely originating from a duplication event of this chromosomal region. Indeed, a highly homologous 5.7-kb pseudogene (GBAP1 GRCh37/hg19 Chromosome 1:155,183,616 to 155,197,325) is located approximately 16 kb downstream of the functional GBA1 gene1. The high degree of homology, which reaches 96% in exoninc regions, and the proximity between GBA1 and GBAP1 enables the occurrence of recombination events resulting in complex gene-pseudogene rearrangements. Indeed, several disease-causing alleles arise from GBA1-GBAP1 rearrangements making the molecular analysis of GBA1 challenging. Therefore, adequate strategies should be adopted to avoid misdetection of GBA1 variants2.
Historically, GBA1 variants were numbered considering the first residue after the cleavage of the signal peptide as amino acid number one. This nomenclature is still used (here reported between brackets and without the prefix p.), although it does not comply with nomenclature standards of the Human Genome Variation Society (HGVS).
To date, over 500 variants in the GBA1 gene have been reported (HGMD Professional). Although most of them are missense, all kind of defects such as splicing alterations, insertions, partial and total deletions, gene-pseudogene rearrangements have been reported. It is worth noting that some variants have never been reported in GD patients but have been identified as associated to Parkinson disease.
A striking difference in distribution of variants has been observed in different populations. Mutations pN409S (N370S), c.84dupG, p.L483P (L444P), c.115+1G>A account for 90% of pathogenic alleles in the Jewish population whereas they represent fewer than 75% of alleles among non-Jewish Caucasian patients. Despite these differences, pN409S (N370S) and p.L483P (L444P) remain the most frequent disease associated variants identified world-wide3.
Genotype-phenotype correlation is poor for most GBA1 mutations with some exceptions.
Patients carrying the pN409S (N370S) variant in at least one allele do not develop primary neurological disease. However, patients with this variant are still at risk for Parkinson disease. While homozygous patients present a mild phenotype or are even asymptomatic, the clinical picture and the severity of the diseases is quite heterogeneous in compound heterozygous individuals.
The correlation is more complex for the second most frequent variant; pL483P (L444P). Indeed, homozygosity for this variant has been associated with the neurological phenotype. However, a wide phenotypic variability has been observed in these patients and several individuals (including adults) with this genotype and without neurologic involvement have been reported. Variants p.G241R (G202R), p.F252I (F213I) and p.R170C (R131C) are strongly associated with the neurological phenotype3.
The p.N227S (N188S) variant, initially classified as a mild mutation associated with type 1 GD, is indeed associated with myoclonic epilepsy4.
Individuals homozygous for the p.D448H (D409H) allele present with an atypical phenotype (type 3C GD) characterized by cardiovascular disease with calcification of the mitral and aortic valves, aortic calcification, hydrocephalus and corneal opacity .It is important to highlight that the presence in cis of the non-pathogenic variant p.H294Q (H255Q) leads to a more severe neurological phenotype5. These findings stress the need to adopt adequate strategies to ensure correct patient’s genotyping of this particularly complex locus. Indeed, the presence of mutations in cis as part of complex or recombinant alleles is not infrequent and can modify the clinical outcome.
A founder effect for specific alleles has been described in different populations, such us the p.N409S (N370S) in Ashkenazi Jewish, Spanish, and Portuguese patients; the p.L483P (L444P) in Swedish and the complex allele [p.D448H (D409H); p.H294Q (H255Q)] in subjects from the Balkans and Greece.
Genetic counselling
Genetic counselling must be offered to all newly diagnosed patients and family members to provide information about the genetic testing, mode of inheritance and risk of recurrence of the disease and in order to promote informed choices.
Once GBA1 pathogenic variants are identified in a proband, genetic testing can be offer to at risk family members. It is important to point out that genetic testing is the only reliable tool to identify heterozygous carriers.
The offspring of GD patients are obligate heterozygotes (carriers) for pathogenic variants in the GBA1 gene while, in most cases, both parents of affected patients are carriers.
These couples in which both partners are heterozygous carriers have in each pregnancy, a 25% chance of conceiving an affected child, a 50% chance of conceiving an asymptomatic carrier, and a 25% chance of conceiving an unaffected and not a carrier. Therefore, once the carrier status of both partners has been confirmed, prenatal or preimplantation genetic testing for diagnosis for GD can be offered.
Genetic testing of reproductive partners of affected patients or heterozygous carriers should be considered in certain populations in which the carrier frequency is high. In the Ashkenazi Jewish population, for example, one in 18 individuals is a carrier for GD, increasing the risk that an affected individual or a healthy carrier may have a reproductive partner who is heterozygous.
References:
- Horowitz M, Wilder S, Horowitz Z, et al. The human glucocerebrosidase gene and pseudogene: structure and evolution. Genomics. 1989;4(1):87–96.
- Zampieri S, Cattarossi S, Bembi B, Dardis A. GBA Analysis in Next-Generation Era: Pitfalls, Challenges, and Possible Solutions. J Mol Diagn. 2017;19(5):733–741.
- Hruska KS, LaMarca ME, Scott CR, Sidransky E. Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum. Mutat. 2008;29(5):567–583.
- Kowarz L, Goker-Alpan O, Banerjee-Basu S, et al. Gaucher mutation N188S is associated with myoclonic epilepsy. Hum. Mutat. 2005;26(3):271–273; author reply 274-275.
- Santamaria R, Michelakakis H, Moraitou M, et al. Haplotype analysis suggests a single Balkan origin for the Gaucher disease [D409H;H255Q] double mutant allele. Hum. Mutat. 2008;29(6):E58-67.
Symptomatic treatment
Prior to the availability of enzyme replacement therapy (ERT) in the early 1990s, the treatment of type 1 Gaucher disease (GD) was limited to symptom control. Massive splenomegaly, causing mechanical symptoms or severe anaemia and thrombocytopenia, sometimes necessitated removal of the spleen. This generally led to complete hematological recovery, and especially of platelets. Unfortunately, splenectomy has been shown to be associated with increased severity of Gaucher-related bone disease, and increased incidence of gall stones, and pulmonary complications and malignancy. Partial splenectomy has also been attempted to avoid worsening of the disease, but regrowth or the spleen and surgical complications have diminished its use. Splenectomy can now almost always be avoided by the institution of ERT. In rare instances, for example when there is severe fibrosis, ERT may fail to reduce the size of the spleen, and splenectomy for persistent hypersplenism may still be necessary. Symptomatic therapy of bone crises includes analgesic drugs and bed rest. Attempts should be made to differentiate the clinical features of a bone crises from bacterial osteomyelitis, which would require lengthy treatments with intravenous antibiotics. Orthopedic procedures can be indicated when there are pathological fractures or where avascular necrosis results in secondary arthritis and joint destruction. Other elements of supportive care include optimization of bone density, provision of blood products where indicated and evaluation and support of bleeding tendency prior to surgical intervention or parturition.
Enzyme replacement therapy
Early studies of ERT involved either the intravenous infusion of small amounts of purified, unmodified glucocerebrosidase (GCase) from placental tissue, or the administration of enzyme entrapped in liposomes or erythrocyte ghosts to facilitate macrophage uptake. Improved targeting of the enzyme to macrophages was ultimately achieved (by modification of its glycosylation status and exposure of terminal mannose residues. Clinical trials with this modified enzyme demonstrated an important clinical effect1. The industrial scale production of placenta-derived GCase (Alglucerase) and the development of the recombinant enzyme, Imiglucerase (Ceredase® and Cerezyme®), made possible several further clinical trials, which confirmed the beneficial effects of enzyme therapy. In the last decade two other ERTs have become available for GD: velaglucerase alfa by gene activation technology in a human cell line (Vpriv®), and taliglucerase alpha, plant cell-based protein expression in carrot cells (Elelyso®). The protein sequence of velaglucerase differs from imiglucerase and taliglucerase in that it is the native human sequence whereas in the others there is a histidine for arginine amino acid substitution at position 495. The three enzymes are not biosimilar, and the availability may differ between different countries. Recently, imiglucerase biosimilars and bioanalogues have been approved (Abcertin™, Glurasim®).
The high cost of ERT has resulted in variation of dosing regimen with consideration of lower dose. In addition, treatment is of limited availability in developing countries, often through charitable access schemes. Initial studies were performed with 60U/Kg every 2 weeks and whilst this dose is commonly used in some countries other investigators advocate a personalized dosing regimen of lower doses tailored to the response of a number of clinical and pathological outcome measures (therapeutic goals)2. Furthermore, home treatment, has been shown to be feasible, convenient and cost-effective3. In the Netherlands, a protocol was developed that allowed individualized treatment based upon each patient’s response, potentially providing an improved cost–benefit ratio4. In most patients, improvement in cytopenia and decreases in splenic and hepatic size are apparent after 3–12 months of treatment. Improvements in the hemoglobin level and especially in the platelet count are usually faster in splenectomised than in non-splenectomised patients. Liver volumes usually normalize, while some degree of splenic enlargement commonly persists, even after long-term treatment. Retarded growth in children and quality of life improves with treatment. Bone disease tends to respond much less rapidly than organomegaly to ERT, but this depends largely on the sensitivity of the methods employed to assess the response.
ERT is generally well tolerated with a low incidence of infusion reactions. The prevalence of IgG antibodies in patients receiving enzyme infusions differs by enzyme product but is not consistently associated with reduced therapeutic effect.
Despite the great success of ERT, several issues remain unresolved. For example, despite local guidelines there remains no consensus on the criteria for initiation of treatment, the best way to monitor effects, and the most appropriate dosing regimen, at particularly during the maintenance phases of therapy. Therapeutic goals can be defined for individuals with the use of clinically relevant and personalized therapeutic endpoints however protocols could be developed to reduce variation in practice and potentially outcomes. Intravenously infused recombinant enzyme does not cross the blood brain barrier and therefore has limited impact on the central nervous system in neuronopathic forms of GD however somatic disease responds well and ERT is indicated for improvement of hematological manifestations, organ volumes and bone disease.
Substrate inhibition therapy
The accumulation of glucosylceramide is due to an imbalance between the rates of its synthesis and degradation. This concept provides the underlying basis for an alternative oral therapy: substrate inhibition therapy (also known as substrate reduction therapy (SRT). Clearance of the accumulated glycolipid should be possible by attenuating the rate of synthesis of the substrate to a level that is matched to the residual activity of the endogenous, mutant glucosylceramidase. N-butyldeoxynojirimycin (miglustat) is an inhibitor of glucosylceramide synthesis, the first committed step in the biosynthesis of glycolipids. This compound which reduces the amount of storage material, resulted in improvements in all key clinical features and biochemical abnormalities in clinical trials of patients with type 1 GD5. Liver and spleen volumes showed a gradual decrease in the first 6 months of treatment, while hematological improvement was slower. However, compared to ERT, the effects of miglustat were slower to manifest and side effects including gastrointestinal disturbance and occasional peripheral neuropathy were noted. The drug has been used in patients with mild to moderate GD in who ERT was not possible. More recently another SRT, eliglustat, has been approved in the US and EU for treatment of adults with type 1 GD (GD1)6. In a randomized trial of 40 untreated GD1 adults therapy with eliglustat for nine months lead to reductions in spleen and liver volume, and improvements of platelet count, and haemoglobin compared with placebo. In a study of patients randomised to switch to eliglustat from ERT or continue with ERT, eliglustat was noninferior to imiglucerase in the composite endpoint of decreased hematologic measurements (hemoglobin and platelet count) and increased organ volume (spleen and liver). Eliglustat is metabolised through cytochrome p450 (CYP2D6) and therefore dosing must consider metabolizer status, with extensive and intermediate metabolizers receiving 84 mg orally twice daily, poor metabolizers 84 mg once daily and in CYP2D6 ultra-rapid metabolizers the drug is contraindicated. Eliglustat has a distinct side effect profile compared to ERT and is contraindicated in pregnancy, lactation, patients with exciting cardiac disease, arrhythmia, renal, and liver disease and not yet approved for children with GD. Similar to ERT, eliglustat cannot cross the blood-brain barrier and thus, similar to ERT, has limited impact on the central nervous system in neuronopathic forms of GD and should be targeted to adult patients with GD1 only; a clinical trial of eliglustat in children is undergoing. A new SRT, venglustat, that crosses the blood-brain barrier, is currently in phase 2 clinical trials for GD37.
Other potential therapies
Haematopoietic stem cell transplantation has been used to good effect in patients with neuronopathic and non-neuronopathic GD but is associated with risk of side effects and procedure- associated mortality. It has now therefore largely been superseded using ERT or SRT. Enzyme enhancement or pharmacological chaperone therapy is the use of a small molecule to improve the stability of endogenous enzyme and therefore its availability in the lysosome. Ambroxol is a mucolytic which is available in some countries, but which, in addition, has been demonstrated in vitro to have pharmacological chaperone activity for GCase in vitro and in a few small studies has been associated with neurological improvement in patients with neuronopathic GD8. Further confirmatory studies are required. Finally, gene therapy approaches to sustained delivery of ERT with either in vivo strategies using adenovirus vectors or ex vivo using lentivirus in stem cells are in development.
In general, patients with rare disease, such as GD, should be managed by a multidisciplinary team with long term experience with the diverse clinical manifestation and GD-related and unrelated com-morbidities9. Disease severity scoring systems for adults and children with GD should be used to classify disease severity, monitoring disease progression, making decisions about when to treat, and monitoring disease improvement with therapy. Patient-reported outcomes measures (PROMs) as a way to ascertain patients with GD views of their symptoms, their functional status, and their health-related quality-of-life are currently in development.
References
- Barton NW, Brady RO, Dambrosia JM, et al. Replacement therapy for inherited enzyme deficiency–macrophage-targeted glucocerebrosidase for Gaucher’s disease. N. Engl. J. Med. 1991;324(21):1464–1470.
- Biegstraaten M, Cox TM, Belmatoug N, et al. Management goals for type 1 Gaucher disease: An expert consensus document from the European working group on Gaucher disease. Blood Cells Mol. Dis. 2018;68:203–208.
- Zimran A, Hollak CE, Abrahamov A, et al. Home treatment with intravenous enzyme replacement therapy for Gaucher disease: an international collaborative study of 33 patients. Blood. 1993;82(4):1107–1109.
- Hollak CE, Aerts JM, Goudsmit R, et al. Individualised low-dose alglucerase therapy for type 1 Gaucher’s disease. Lancet. 1995;345(8963):1474–1478.
- Cox T, Lachmann R, Hollak C, et al. Novel oral treatment of Gaucher’s disease with N-butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis. Lancet. 2000;355(9214):1481–1485.
- Mistry PK, Balwani M, Baris HN, et al. Safety, efficacy, and authorization of eliglustat as a first-line therapy in Gaucher disease type 1. Blood Cells Mol. Dis. 2018;71:71–74.
- Schiffmann R, Mengel E, Carwford N, et al. Venglustat in adult Gaucher disease type 3: Preliminary safety, pharmacology, and exploratory efficacy from a phase 2 trial in combination with imiglucerase (LEAP). Mol Gen Metab. 2019;126:S131.
- Narita A, Shirai K, Itamura S, et al. Ambroxol chaperone therapy for neuronopathic Gaucher disease: A pilot study. Ann Clin Transl Neurol. 2016;3(3):200–215.
- Revel-Vilk S, Szer J, Mehta A, Zimran A. How we manage Gaucher Disease in the era of choices. Br. J. Haematol. 2018;182(4):467–480.
Philippe Gaucher
COMING SOON
Diagnosis
A diagnosis of Gaucher disease is usually based upon histological or cytological examination of bone marrow specimens, a liver biopsy, or a surgically removed spleen. The presence of lipid-laden macrophages with striated cytoplasmic inclusions, the Gaucher-cells, is not specific for the disease. Many other diseases, including chronic myeloproliferative diseases, malignancies, and chronic inflammatory disorders, have been described as giving rise to so-called ‘pseudo-Gaucher’ cells. For a reliable diagnosis of Gaucher disease, histological examinations are neither necessary nor sufficient. The detection of low glucocerebrosidase levels in leukocytes or urine is pathognomonic of Gaucher disease and should therefore be applied in cases of suspected Gaucher disease. However, since the disorder is rare and its clinical manifestations may mimic lymphoma or other haematological diseases, a bone marrow or liver biopsy will usually be performed before a diagnosis of Gaucher disease is even considered, especially in the absence of known affected family members. For the detection of carrier status, enzyme assays have limited value because of the considerable overlap with normal subjects. In these cases, mutation analysis is warranted.
Heredity
Gaucher disease is an autosomal recessively inherited disorder caused by mutations of the GBA gene. At present, more than 200 mutations have been established (http://www.tau.ac.il/racheli/genedis/gaucher/gaucher.html).
The gene, which has been mapped to chromosome 1q21, is comprised of 11 exons ( 7 kb) and encoded by a cDNA strand of about 2.5 kb. There are two upstream start codons (ATG) that are both utilized in translation. A highly homologous pseudogene ( 5 kb) showing 96% exonic sequence similarity is located 16 kb downstream. The GBA gene mutations are numbered according to their position in the cDNA relative to the upstream ATG codon (where the A is designated 1), or identified on the basis of the amino acid position in the protein sequence. The abnormal alleles include exonic missense and nonsense mutations, splice junction mutations, deletions or insertions of one or more nucleotides, and complex alleles resulting from gene conversion or recombination. Four mutant alleles (N370S, L444P, 84GG, IVS2+1) account for the majority of disease-causing alleles. Screening studies involving Ashkenazi Jews revealed that the two most common mutations, N370S and 84GG, have a frequency of 0.032 and 0.00217, respectively, and account for approximately 77% and 13%, respectively, of the mutant alleles found in this population.
Genotype–phenotype correlations are imperfect, and the wide variability in clinical presentations among Gaucher disease patients represents a major confounding issue in genetic counselling. The common mutations have been associated with various clinical subtypes, although the presence of at least one N370S allele appears to preclude the development of neuropathic involvement.
Laboratory features
Glucocerebrosidase is the lysosomal hydrolase responsible for the degradation of the natural glycosphingolipid, glucosylceramide (glucocerebroside), into ceramide and glucose. Deficiency of this enzyme results in the accumulation of undegraded glucosylceramide, almost exclusively in macrophages. The glucosylceramide concentration in the spleen can be increased 10- to 1000-fold, but high levels are also detected in liver and bone marrow specimens. In contrast, the plasma concentration of glucosylceramide is only moderately increased, on average approximately two-fold.
Significant decreases or downward trends in glucosylceramide concentrations in response to enzyme therapy have been noted in patients with various Gaucher disease phenotypes. Apart from glycolipid levels, a considerable number of other measurements change. Abnormalities in the levels of other proteins, including acid hydrolases and lipids, have been observed in the tissues and plasma of Gaucher patients. Several of these abnormalities are of interest because of their potential role in the pathophysiology of the disease, their ability to serve as markers for disease severity, and their application in measuring the efficacy of therapeutic intervention. One of the earliest examples is the elevated plasma concentration of tartrate-resistant acid phosphatase 5B (TRAP), which is secreted by the Gaucher macrophages. Other plasma abnormalities frequently used to monitor disease progression and response to therapy are ferritin, angiotensin-converting enzyme (ACE), hexosaminidase, and the lysosomal hydrolase, chitotriosidase.
The latter has been discovered by AMC researchers and is by far the most highly elevated enzyme in symptomatic patients. In clinically affected individuals, chitotriosidase levels are elevated at least 100-fold, but may be higher than 600 times the median value detected in healthy volunteers [1]. The function of chitotriosidase is as yet unknown, but its close resemblance to the non-vertebrate chitinases, which act in the defence against or degradation of chitin-containing pathogens, such as fungi, may provide a clue to its biological role in humans. Extensive studies have revealed that chitotriosidase originates from the Gaucher cell and has a close relationship with the body burden of Gaucher cells. It can thus be used in addition to other parameters of disease activity to monitor disease progression and response to therapy. However, one has to bear in mind that 6% of the entire population, including Gaucher patients, have a complete lack of plasma chitotriosidase activity due to a single mutation in the chitotriosidase gene [2]. The carrier prevalence for this mutation can be as high as 40%, resulting in lower levels of chitotriosidase activity. CCL18/PARC is a chemokine secreted by Gaucher cells as well and can be used as a reliable alternative, although elevations are not as striking as found for chitotriosidase. More recent studies revealed elevations in MIP1beta. This cytokine is thought to play a role in bone remodelling and may therefore be a better marker for ongoing bone disease symptomatology. Several other plasma abnormalities described in Gaucher disease may contribute to the clinical picture, including reduced lipoprotein levels, hypergammaglobulinaemia, and elevated levels of proinflammatory cytokines and macrophage-activation markers.
Therapy
Palliative treatment
Before enzyme replacement therapy became available in the early 1990s, the treatment of type 1 Gaucher disease was merely palliative. Massive splenomegaly, causing severe anaemia and thrombocytopenia or leading to mechanical complaints, sometimes necessitated removal of the spleen. This almost invariably led to complete haematological recovery, especially of platelet counts. Whether splenectomy results in progressive storage of glycolipids at other sites, such as the liver, lung and bone marrow, has been subject to debate. Partial splenectomy has also been attempted to avoid the removal of the spleen as a major storage site, but splenic regrowth and surgical complications abandoned its use. In the majority of cases, splenectomy can be avoided with the institution of enzyme therapy. In rare instances, e.g. the presence of severe fibrosis, enzyme therapy may fail to reduce the size of the spleen, and splenectomy for persistent hypersplenism may still be necessary. For the palliative treatment of bone crises, analgesic drugs and bed rest are usually indicated. Bacterial osteomyelitis, which can be difficult to distinguish from bone crises, requires lengthy treatments with intravenous antibiotics. Orthopaedic procedures are sometimes indicated in cases of avascular necrosis or pathological fractures.
Enzyme replacement therapy
Early studies of enzyme replacement therapy involved either the intravenous infusion of small amounts of purified, unmodified glucocerebrosidase from placental tissue, or the administration of enzyme entrapped in liposomes or erythrocyte ghosts to facilitate macrophage uptake. The results of these studies were disappointing. However, improved targeting of the enzyme to macrophages was achieved (by modification of its glycosylation status and exposure of terminal mannose residues) and higher doses used. The first clinical trials with this new formulation showed a dramatic response in patients to regular intravenous infusion of this modified enzyme [1]. The industrial scale production of placenta-derived glucocerebrosidase and the development of the recombinant enzyme (CeredaseTM and CerezymeTM) made possible a number of further clinical trials, which confirmed the beneficial effects of enzyme therapy. Extremely high costs have made this treatment available only for those living in developed countries, and lower dosing regimens have thus been considered. Initial studies were performed with bi-weekly infusions of 60 U/kg per month. Other investigators advocated that lower dosages at higher frequencies were more efficient and therefore more cost-effective. In addition, home treatment, which further lowers costs, was shown to be feasible and more convenient for the patients [2]. In the Netherlands, a protocol was developed that allowed individualized treatment based upon each patient’s response, potentially providing an improved cost–benefit ratio [3]. In most patients, improvement in cytopenia and decreases in splenic and hepatic size are apparent after 3–12 months of treatment. Improvements in the haemoglobin level and especially in the platelet count are usually faster in splenectomised than in non-splenectomised patients. Liver volumes usually normalize, while some degree of splenic enlargement commonly persists, even after long-term treatment. Retarded growth in children and quality of life improves with treatment. Bone disease tends to respond much less rapidly than organomegaly to enzyme replacement therapy, but this depends largely on the sensitivity of the methods employed to assess the response. Many tools have been applied to assess organ responses to enzyme replacement. Plain X-rays are very insensitive and have no place in the assessment of treatment response; they should only be used in cases of bone complications. Magnetic resonance imaging is probably the best way to obtain information on bone marrow invasion and structure. Several scoring systems have been developed based upon changes in T1- and T2-weighted patterns. Quantitative chemical-shift imaging (QCSI) is the most sensitive method to assess bone marrow infiltration. This technique was further developed at the AMC by Mario Maas and Erik Akkerman and measures the ratio of triglyceride to water in the bone marrow [4]. In Gaucher disease patients, this ratio is greatly reduced, probably due to displacement of normal triglyceride-rich adipocytes in the bone marrow by Gaucher cells. AMC researchers have shown that the fat fraction of the bone marrow at the level of the lumbar spine relates to bone complications. Early reappearance of fatty marrow during enzyme replacement therapy can be detected with this technique. However, QCSI is not widely available and therefore limited in its use.
Despite the enormous success of enzyme therapy, several issues remain unresolved. For example, there is still no consensus about the criteria for initiation of treatment, the best way to monitor effects, and the most appropriate dosing regimen, particularly during the maintenance phase of treatment. The variability in clinical response to treatment and the extremely high costs (around $ 200 000 to 500 000 per patient per year) play an important role in these issues. As commitment to therapy is potentially life-long, the overall cost of care using enzyme therapy can be considerable, and very few reports are available about long-term therapeutic outcomes. Generally, therapeutic goals should be defined with the use of clinically relevant therapeutic endpoints, and protocols should be developed that aim to maintain an optimal effect while decreasing the burden of frequent infusions and the cost of care.
Substrate balance therapy
The accumulation of glucosylceramide is due to an imbalance between the rates of its synthesis and degradation. This concept forms the basis of a recently developed alternative oral therapy, termed substrate balance therapy (also known as substrate reduction therapy). Clearance of the accumulated glycolipid should be possible by attenuating the rate of synthesis of the substrate to a level that is matched to the residual activity of the endogenous, mutant glucosylceramidase. N-butyldeoxynojirimycin (OGT918 or miglustat) is an inhibitor of glucosylceramide synthesis, the first committed step in the biosynthesis of glycolipids. When tested in animal models of glycolipid storage disorders, the compound reduces the amount of storage material and delays the onset and progression of disease manifestations. Its mode of action suggests therapeutic potential in several of these diseases, including those with neurological involvement, since this small molecule is capable of crossing the blood–brain barrier. The first study in patients with a lysosomal storage disorder was initiated in 1998 in 28 type 1 Gaucher disease patients, who received 100 mg OGT918 orally three times daily [5]. The results of this study were promising: improvements occurred for all key clinical features and biochemical abnormalities. Liver and spleen volumes showed a gradual decrease in the first 6 months of treatment, while haematological improvement was slower. The most common adverse event was diarrhoea, which occurred in almost 90% of patients, especially during the first 3 months of treatment. Compared to enzyme therapy, the effects of OGT918 are slower to manifest. This and the disadvantageous side effects have to be balanced against the advantages of oral administration. Enzyme therapy remains the first choice for patients with moderate to severe disease, but mildly affected patients or those with minimal residual disease after treatment with enzyme may prefer this oral alternative. New oral alternatives are currently being developed.